Table Of Content
Moreover, the researchers have published the algorithm and its software so that researchers worldwide can now use them for their own projects. Dr. Julia Schaletzky is the Founder of the UCB Drug Discovery Center, the Executive Director of the Center for Emerging and Neglected Diseases, as well as of the Immunotherapy and Vaccine Research Initiative at UC Berkeley. An expert in drug discovery and preclinical development, Dr. Schaletzky has more than 10 years industry experience, contributing to the development of first-in-class therapies for heart failure and neurodegenerative diseases. The UC DDC is governed by a diverse group of experts in drug discovery and development that serve as site lead representatives for their respective UC campuses. The researchers aren't now pursuing these molecules any further with a view to bringing drugs based on them to the market.
University of Southern California
These results highlight their ability to target PPAR with precision, while also sidestepping pronounced influence on closely affiliated nuclear hormone receptors, like RXR, and a sizeable panel of other undesired off-targets. This outcome demonstrates the efficacy of the structure-based DRAGONFLY de novo design approach in generating molecules with the desired properties and biological activities, including selected ADME properties. The observed lack of CYP interaction up to a compound concentration of 10 μM is a crucial aspect in averting drug-drug interactions, which is of particular relevance for the treatment of metabolic syndrome, where patients frequently require concurrent administration of multiple drugs71.
AI is boosting drug discovery and development — and sparking questions about proprietary data - GeekWire
AI is boosting drug discovery and development — and sparking questions about proprietary data.
Posted: Fri, 19 Apr 2024 16:10:27 GMT [source]
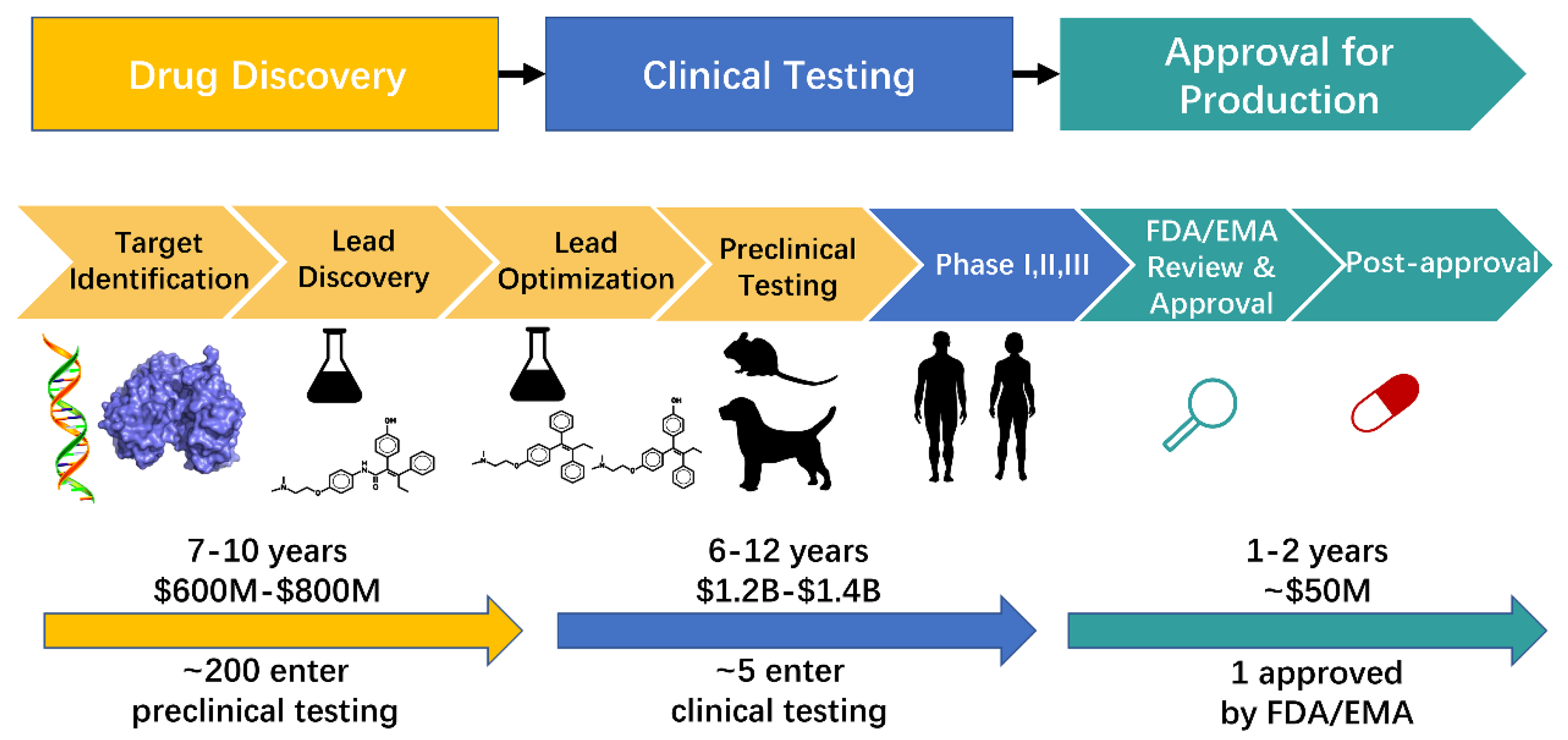
Figures
The developments in the pharmacokinetics of opioids is considered as a case study to briefly show the role of metabolism as a predictor of the clinical response and side effects of opioid analgesics, keeping the opioid crisis in mind [76]. The important side effects are due to the neuronal connectivity between the reward, dopaminergic, and opioid regions, as well as to the respiratory depression in the CNS, while many other side effects, e.g., constipation, are derived from the interaction with the peripheral opioid receptors. The cherry-top of the automation and current application of artificial intelligence in drug design are the robots Adam and Eve, constructed by Ross King and Stephen Oliver from the University of Manchester. Adam was constructed to run microbiological experiments, analyse the results itself, define hypotheses, design experiments to test these hypotheses and repeat this cycle until a validated hypothesis is derived [61].
Assessing sequence-based protein–protein interaction predictors for use in therapeutic peptide engineering
Founded in 1829 as a private technical institute, the curriculum at RIT emphasizes 'career education and experiential learning'. Undergraduate programs prepare students for careers in areas such as Pathology, Pharmacy, Pharmacology and Drug Development, Toxicology, Neuroscience and Genetic Counseling. Dr. Robert Vince, medicinal chemist, researcher, and holder of over 20 medical patents, founded the Center for Drug Design at the University of Minnesota in 2002. Now the director of the center, one of Dr. Vince's most notable achievements was the design of an anti-HIV/AIDS medicine called "carbovir".
Again, DL has proven useful in aiding the fine tailoring of the best treatment choice based on the analysis of patient data such as life history, previous diagnostics, and manifested symptoms [53]. De novo drug design aims to generate molecules from scratch that possess specific chemical and pharmacological properties. We present a computational approach utilizing interactome-based deep learning for ligand- and structure-based generation of drug-like molecules.
How did your time at the USC Mann School impact you?
Additionally, staff at a rehabilitation center can teach you the coping skills for managing post-rehab life so you don’t relapse and fall back into your old addiction. Designer drugs can be incredibly dangerous and carry risks of serious health problems, fines and arrests, and even the potential of overdosing. Let’s take a closer look at some common designer drug examples and their potential side effects. For example, some of these drugs are sold and advertised as legal simply because they’re labeled as not for human consumption. Alternatively, a person may think they’re taking a normal street drug and not realize they’ve been given a designer drug version.
It operates on diverse chemical alphabets and does not require fine-tuning through transfer or reinforcement learning specific to a particular application. Furthermore, it enables the incorporation of desired physical and chemical properties into the generation of output molecules. This study introduces the prospective application of DRAGONFLY to structure-based de novo design, specifically for the generation of ligands with desired bioactivity profiles addressing one or multiple specific macromolecular targets (Fig. 1f). Protein structure-based drug development has been a successful approach for diseases with well-defined protein targets over the past few decades1,2,3. A typical protein structure-based drug design (SBDD) project starts from the protein sequence and builds a three-dimensional (3D) structure through structural biology or structure prediction. It then identifies binding pockets, including orthosteric sites or allosteric sites, and finally discovers active modulators through virtual screening or de novo design4,5 (Fig. 1a).

This puts the pharma industry in the top three most profitable industries in the world. Synthetic drugs carry extra risk because you can’t be sure exactly what you’re consuming, injecting or smoking, so there’s no way of knowing how your body will respond when you try to stop using them. A rehabilitation center can provide 24/7 monitoring, so if any health issues arise, medical staff is on standby ready to help.
The test compound was titrated into buffer, and the buffer was titrated to the PPARγ LBD proteins under otherwise identical conditions. The ITC results were analyzed using NanoAnalyze software (TA Instruments, New Castle, DE) with an independent binding model. The isolated CT26 tumor tissue was fixed with neutral paraformaldehyde, and subsequent staining of cell surface markers was performed by Servicebio Company (Wuhan, China). In brief, the tumor tissue embedded in paraffin was processed through sectioning, dewaxing, rehydration, and antigen retrieval. Following peroxidase inactivation and blocking with goat serum, the tissue was incubated overnight with the corresponding primary monoclonal antibody overnight at 4 °C.
Through docking-based virtual screening, hit structures can be identified among huge datasets binding to a given binding site [20]. The pharmacophore is a 3D ensample of functional groups in the active molecules necessary for binding to a given receptor. For the best performance, both approaches for virtual screening are complimentarily used [59,60]. Once there is a quantitative description of a set of structures and a quantitative measurement of their activities such as IC50, EC50, Kd, etc., then different types of quantitative analyses can be applied. Corwin Hansch conducted this for the first time in 1964 when correlating the antimicrobial activity of penicillin derivatives with descriptors relating to the hydrophobic and electronic properties of the molecules [33].
The resulting subset of molecules obtained from the filtering process was further ranked using KRR-based QSAR scoring based on the average predicted binding affinity (pKI or pIC50), combining the ECFP (double weighted contribution), CATS, and USRCAT descriptors. Aiming to explore the potential of the computer-generated molecules for dual-target activity and receptor selectivity, two different scoring procedures were prospectively evaluated. The second procedure involved assigning equal weights to dual-target affinity towards both PPARγ and PPARδ. The decision to focus on affinity towards PPAR sub-family members was made to align with their clinical significance48.
The starting point and the data accumulated in a given project will be treated in a global perspective and will generate an unexpected synergy between projects that may appear unrelated. In the 1980's, protein homology models were constructed to compensate for the lack of X-ray data in order to provide data for direct drug design of new molecules. When information is available for both the target protein and active molecules, the two approaches can be developed independently. In the first case the design will concentrate on the binding to the 3D structure of the protein, and in the second case it will be based on the structures of the reference active molecules. When the three-dimensional structure of the target protein is known, the receptor-based design exploits the recognition and discrimination capabilities of the receptor site to create direct interactions between the designed molecule and atoms or functional groups of the target protein.
A protein thermal shift assay (PTS) revealed dose-dependent Tm shifts (Supplementary Fig. 2a), indicating that 230D7 could bind directly to SPOPMATH. Additionally, NMR experiments confirmed the direct binding between SPOPMATH and 230D7 (Supplementary Fig. 2b). An in vitro pull-down assay was performed to verify that 230D7 dose-dependently reduces PTEN binding to SPOPMATH (Supplementary Fig. 2c, d). After validating the molecular activity, we used 230D7 for the functional study at the cellular level.
A recent example that explicitly considers the time evolution of a target molecule is the PPI-FIT method, which involves the targeting of intermediates along the path of protein folding (Figure 3) [21]. These structures are regarded as the druggable targets because they present binding pockets not present in the protein’s final structure. The drug-intermediate interaction should stabilize the complex, thus preventing the protein from reaching its native conformation. The method employs computer simulations together with experimental techniques, and supports the idea that folding intermediate targeting could represent a useful way to regulate protein levels. Working on a single crystal is marginally useful to understanding the enzyme movements during the catalytic process and to plan possible molecular structures interacting or interfering with different conformational states of the enzyme.
For histological analysis of BALB/c mice in 230D7-treated or vehicle control groups, H&E staining were performed using standard histological techniques. According to the manufacturer’s protocol (Servicebio, Inc.), isolated organ tissues were fixed in 4% neutral paraformaldehyde for 24 h and embedded in paraffin wax. Lastly, the slides were sealed with neutral resin and images were captured by microscopy (Eclipse E100, DS-U3, Nikon).








No comments:
Post a Comment