Table Of Content
- Professional development
- Types of CADD
- The transformational role of GPU computing and deep learning in drug discovery
- Computer Aided Drug Design and its Application to the Development of Potential Drugs for Neurodegenerative Disorders
- Modular synthon-based approaches
- Further growth of readily accessible chemical spaces
Second, the accessibility of hit analogues in the same on-demand spaces streamlines a generation of meaningful structure–activity relationship (SAR)-by-catalogue and further optimization steps, reducing the amount of elaborate custom synthesis. Last, although the library scale is important, properly constructed gigascale libraries can expand chemical diversity (even with a few chemical reactions32), chemical novelty and patentability of the hits, as almost all on-demand compounds have never been synthesized before. In silico screening by docking molecules of the virtual library into a receptor structure and predicting its ‘binding score’ is a well-established approach to hit and lead discovery and had a key role in recent drug discovery success stories11,17,51. The docking procedure itself can use molecular mechanics, often in internal coordinate representation, for rapid conformational sampling of fully flexible ligands52,53, using empirical 3D shape-matching approaches54,55, or combining them in a hybrid docking funnel56,57. Special attention is devoted to ligand scoring functions, which are designed to reliably remove non-binders to minimize false-positive predictions, which is especially relevant with the growth of library size. Blind assessments of the performance of structure-based algorithms have been routinely performed as a D3R Grand Challenge community effort58,59, showing continuous improvements in ligand pose and binding energy predictions for the best algorithms.
Professional development
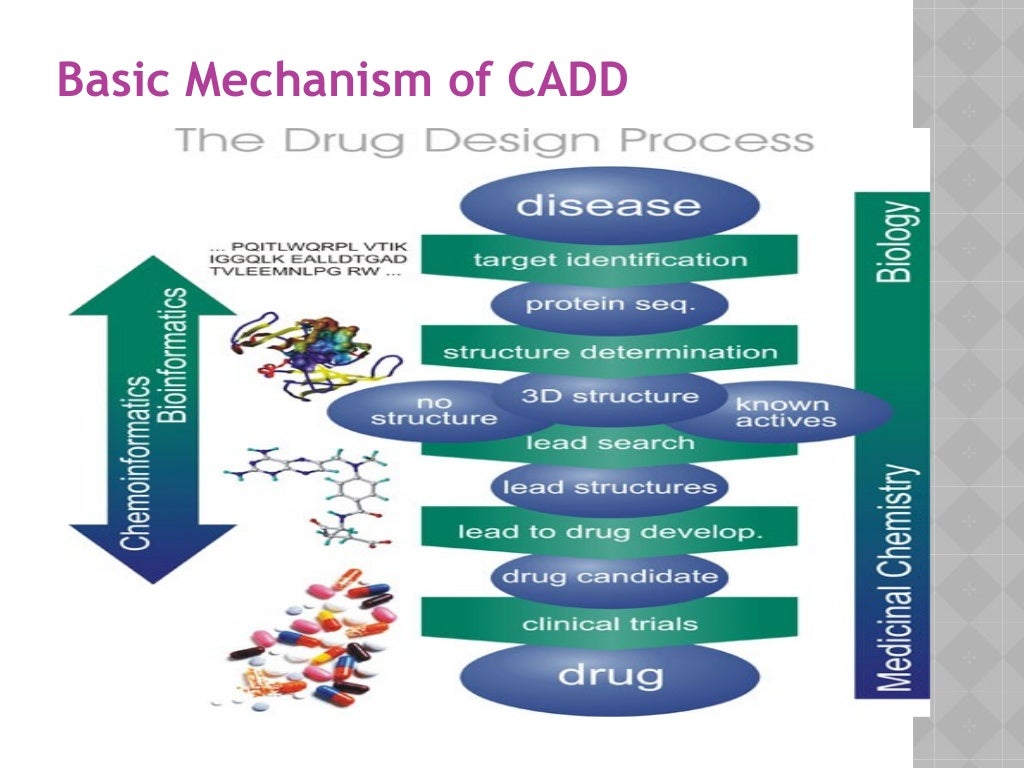
These methods find numerous applications such as assessment of binding energetics, protein-ligand interactions, and conformational changes in the receptor upon binding with a ligand [20]. Being used by many pharmaceutical industries and medicinal chemists, SBDD as a computational technique has greatly helped in the discovery of several drugs available in the market. The basic steps involved in SBDD consist of the preparation of target structure, identification of the ligand binding site, compound library preparation, molecular docking and scoring functions, molecular dynamic simulation, and binding free energy calculation (Figure 1). The recent outbreak of the deadly coronavirus disease 19 (COVID-19) pandemic poses serious health concerns around the world. The lack of approved drugs or vaccines continues to be a challenge and further necessitates the discovery of new therapeutic molecules.
Types of CADD
CAS and Molecule.one Announce a Strategic Collaboration to Accelerate Drug Discovery - Business Wire
CAS and Molecule.one Announce a Strategic Collaboration to Accelerate Drug Discovery.
Posted: Fri, 11 Aug 2023 07:00:00 GMT [source]
An ideal search algorithm should be fast and effective, and the scoring function must be capable of determining the physicochemical properties of molecules and the thermodynamics of interactions. At the early hit identification stage, the ultra-scale virtual screening approaches, both structure-based and AI-based, are becoming mainstream in providing fast and cost-effective entry points into drug discovery campaigns. At the hit-to-lead stage, the more elaborate potency prediction tools such as free energy perturbation and AI-based QSAR often guide rational optimization of ligand potency.
Aurigene Pharmaceutical Services Ltd. introduces Aurigene.AI, an AI and ML assisted drug discovery platform - Express Computer
Aurigene Pharmaceutical Services Ltd. introduces Aurigene.AI, an AI and ML assisted drug discovery platform.
Posted: Wed, 03 Apr 2024 07:00:00 GMT [source]
The transformational role of GPU computing and deep learning in drug discovery
These patterns can be used qualitatively to direct ligand design and, when converted to free energies, termed grid free energy (GFE) FragMaps (66, 67), used to quantitatively estimate the relative binding affinities of ligands. The detailed protocol based on full MD simulations was described previously in this same book series (68). Here we present an updated protocol based on the use of oscillating μex Grand Canonical Monte Carlo/MD (GCMC/MD) simulations for SILCS (69). The GCMC/MD approach allows for the application of the SILCS method to target systems with deep or occluded pockets such as nuclear receptors and GPCRs (70). In the era of AI-based face recognition, ChatGPT and AlphaFold68, there is enormous interest in applications of data-driven DL approaches across drug discovery, from target identification to lead optimization to translational medicine (as reviewed in refs. 69,70,71). Determining the structure of a target molecule follows the identification of a specific drug target [29].
Thus, the generated databases (GDB) predict compounds that can be made of a specific number of atoms; for example, GDB-17 contained 166.4 billion molecules of up to 17 atoms of C, N, O, S and halogens49, whereas GDB-18 made up of 18 atoms would reach an estimated 1013 compounds38. Other generative approaches based on narrower definitions of chemical spaces are now used in de novo ligand design with DL-based generative chemistry (for example, ref. 50), as discussed below. The applicability of CADD for modeling of the drug considers combinatorial chemistry and bioinformatics which address the major issues including cost and time duration. An important alternative to solve the antibiotic resistance issue is the identification of new antibiotic targets that may represent novel mechanisms essential for bacterial survival. Such findings may help to overcome the resistance of this bacterium to common antibiotics such as methicillin, fluoroquinolones and oxazolidinones. An example of a recently identified novel antibiotic target is the protein heme oxygenase, involved in the metabolism of heme by bacteria as required to access iron (10–12).
Structure-based and ligand-based drug design form two branches of the computer-aided drug discovery process which plays a significant role in the design and identification of drug molecules in reduced time and cost. The increase in the number of positive cases and deaths from COVID-19 and the lack of approved drugs and vaccines continue to be a matter of global health concern which necessitates the urgent discovery of drugs for the prevention and cure of the disease. The structural elucidation of pharmacological targets of SARS-CoV-2 has helped the researchers in the structure-based virtual identification of inhibitors, and the discovery of few lead molecules against COVID-19 has led to the use of scaffolds that can be optimized through ligand-based drug design. Realizing the possible mutability of this RNA virus and the emergence of drug resistance problems, it is, therefore, necessary to take a step further and consider targeting multiple drug targets that will be more effective and might help in overcoming drug resistance barriers. The chemical compound which exhibits biological or pharmacological properties with therapeutic characteristics can be called a lead molecule.

Our laboratory together with de Leeuw and coworkers are continuing the design of novel agents against bacteria cell wall biosynthesis (12, 13). In a recent study, SAR for a series of compounds that have benzothiazole indolene scaffold was pursued targeting the essential bacterial cell wall precursor molecule Lipid II (14). Using MD simulations, we predicted binding free energies and binding modes of Lipid II binders and gained atomic details on the interactions between designed molecules with Lipid II, information that will be useful for further development of antibacterial therapeutics.
Further growth of readily accessible chemical spaces

While the spike glycoprotein is essential for the interaction of the virus with the host cell receptor, the nsps play a major role during the virus life cycle by engaging in the production of subgenomic RNAs [13, 14]. The nonstructural and structural proteins, therefore, offer promising targets for the design and development of antiviral agents against COVID-19 [13]. The lack of effective vaccines or drugs for the treatment of COVID-19 and the high mortality rate necessitates the rapid discovery of novel drugs [15], and computer-aided drug design is believed to be an important tool to achieve the identification of novel therapeutics.
Computer-Aided Drug Design Methods
Under the SILCS framework, we recently put forward a protocol to calculate permeation related resistant factor of a molecule to cross membranes (126) using LGFE energy profile and is described in the following. Previously, we published a chapter in the first edition of this book that was dedicated to an overview of CADD and included information on routinely utilized protocols, especially tools used in our laborotary, towards the design of antibotic theraputics (4). Since then CADD methods have been employed extensively to facilitate the development of novel antibiotics by the computational chemistry community and us for the past five years.
While bacterial membranes are complex environments with multiple transport and pore proteins, it is of utility to estimate the pure membrane permeability of drug candidates during drug discovery as this may contribute to drug bioavailability. Traditionally, potential of mean force (PMF) free-energy profiles for a compound across membrane lipid bilayers are derived using MD simulations (124). The PMF may then be used together with position-specific diffusion coefficient in the inhomogeneous solubility-diffusion equation (125) to derive effective resistivity, which may be inverted into permeability.
Although the impacts of the recent structural revolution17 and computing hardware in drug discovery28 are comprehensively reviewed elsewhere, here we focus on the ongoing expansion of accessible drug-like chemical spaces as well as current developments in computational methods for ligand discovery and optimization. We detail how emerging computational tools applied in gigaspace can facilitate the cost-effective discovery of hundreds or even thousands of highly diverse, potent, target-selective and drug-like ligands for a desired target, and put them in the context of experimental approaches (Table 1). Although the full impact of new computational technologies is only starting to affect clinical development, we suggest that their synergistic combination with experimental testing and validation in the drug discovery ecosystem can markedly improve its efficiency in producing better therapeutics.
ML algorithms that have been extensively used in drug discovery include support vector machine (SVM) [105], Random Forest (RF) [106], and Naive Bayesian (NB) [107]. Few examples of the deep learning methods are convolutional neural network (CNN), deep neural network (DNN), recurrent neural network (RNN), autoencoder, and restricted Boltzmann machine (RBN) [4]. The conventional QSAR methods can efficiently predict simple physicochemical properties such as logP and solubility. However, the QSAR prediction of complex biological properties such as drug efficacy and side effects is often not optimal as the methods use small training sets [108] and has coverage of limited chemical space [109]. The big data generated using high-throughput screening (HTS) techniques are huge challenges to traditional QSAR methods and machine learning techniques [40].
Further steps in optimization of the initial hits obtained from standard screening libraries of less than 10 million compounds, however, usually require expensive custom synthesis of analogues, which has been afforded only in a few published cases20,61. He has contributed to the development of a number of technologies in this area, including the creation and maintenance of BindingDB, the first publicly accessible database of protein-small molecule binding data. Dr. Gilson is currently on the faculty of UC San Diego’s Skaggs School of Pharmacy and Pharmaceutical Sciences.








No comments:
Post a Comment